Over the last years, the amount of software used in the healthcare industry has greatly increased. According to Data Bridge, the global Software as a Medical Device (SaMD) market reached $1.58 billion in 2024 and is projected to hit $6.87 billion by 2032 at a CAGR of 20.13%. This rapid market growth is driven by the adoption of technologies such as AI, IoT, and cloud computing, as well as the need for remote and patient-centered care. Such systems have already changed both how healthcare providers interact with patients and how patients themselves monitor their own health.
But when it comes to medical software development, there are a lot of questions around it related to classifications, regulatory pathways, and design principles. This article will help you find out “what is Software as a Medical Device?” and provide a practical guide on how to develop such a solution in a reliable and compliant manner.

What is Software as a Medical Device (SaMD)?
The definition of Software as a Medical Device (SaMD) varies from country to country since regulatory authorities set their own requirements for software. Let’s look at the definitions used in the biggest markets.
Around the globe, the most commonly used definition of SaMD comes from the International Medical Device Regulators Forum (IMDRF). It defines SaMD as software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.
In the United States, the Food and Drug Administration (FDA) defines SaMD as software intended to perform a medical function, such as diagnosis, monitoring, or prediction, without being part of a medical device.
In the European Union, the term “SaMD” is not used directly. Instead, such software is considered within the context of medical device software (MDSW). The EU Medical Devices Regulation (MDR) and the EU In Vitro Diagnostic Devices Regulation (IVDR) define SaMD as medical device software intended for use, either alone or in combination with other devices, for one or more medical purposes as defined by the regulation.
Other important characteristics of SaMD include:
- It can be considered as a medical device, including software for in vitro diagnostics (IVD) to detect diseases or assess a person’s health status based on laboratory tests (for example, blood, saliva, etc.).
- It can operate on different devices, like computers and smartphones not specifically designed for medical use.
- It can operate without being part of a device to achieve its intended medical purpose.
- It can interact with other medical devices and software.
Examples of software that ARE and NOT SaMD
At first glance, it can be challenging to determine whether software falls under the category of Software as a Medical Device. A simple way to do it is to ask the question, “Does software diagnose, predict, monitor, or treat medical conditions of a patient without being part of a hardware medical device? If yes, it is likely SaMD. If the software merely stores or displays data without influencing medical decisions, it is not considered SaMD. To make this clearer, let’s look at some examples.

SaMD vs. SiMD: what is the difference?
Often, software as a medical device (SaMD) is confused with Software in a Medical Device (SiMD) as both types of software are used in the healthcare industry and can influence patient care. In a nutshell, SaMD is software intended to be used for medical purposes without being part of a hardware medical device, while SiMD is software that is embedded within a physical device to perform medical functions. Understanding the core differences between the two systems can help developers determine how software will be regulated, developed, and tested. To clarify how these categories differ, consider the table below:
| SaMD | SiMD | |
|---|---|---|
| Device dependency | Works independently of any medical hardware | Cannot operate independently as relies on the physical device it is part of |
| Main purpose | Serves specific medical functions, such as diagnosis, treatment, monitoring, etc. | Controls the operation of the physical device |
| Development focus | Focus on software only | Focus on both software and hardware |
| Scalability | High, as it can scale among a lot of devices instantly | Limited, as it tied to hardware production and distribution |
| Regulatory pathway | Software-specific regulations, faster approval process | Combined software-hardware regulations, longer approval process |
| Quality management | Software development lifecycle (IEC 62304) | Hardware QMS + software development lifecycle |
| Platform scope | Multi-platform (cloud, mobile, desktop) | Device-specific, transfer to another device is not possible without modifications |
| Updates | Supports rapid, flexible software updates and iterative improvement; faster updates | Updates usually require device-level hardware or firmware changes; slower updates |
| Risk assessment | Based on software impact on patient health | Based on both software functionality and hardware integration risks |
| Examples | Heart-rate tracking app,medication management app, diabetes management app | Infusion pumps software, MRI control system,anesthesia delivery system |
Note: Not all medical software is classified as SaMD or SiMD in the strict sense. Some software is used for research or administrative tasks. None of these types of software would qualify as SaMD or SiMD, as they do not facilitate the diagnosis, treatment, or management of patient diseases.
SaMD regulations and classifications
Once it has been determined that your software qualifies as a medical device, it is important to define its classification. The class will impact the required regulatory pathway and software approval process. As different countries have different SaMD regulations, so medical device manufacturers must stay informed about them.
SaMD regulations and classifications in the US
In the US, the FDA classifies SaMD into three categories: Class I, Class II, and Class III, based on intended use and level of risk to the patient.
- Class I: low-risk software with minimal potential for harm to patients and/or users. Examples include health tracking apps that provide general wellness data without diagnosing or treating medical conditions.
- Class II: moderate-risk software that supports clinical decisions, collects and transmits data on patients’ health status, or monitors non life-threatening conditions. It requires premarket notification (510(k)) to ensure that the device is safe and similar to an already approved device. If a similar device does not yet exist, the De Novo process is used. Example include applications that collect and send psychological symptoms.
- Class III: high-risk software that is used to sustain life, prevent serious deterioration in health, or diagnose/treat conditions that may be life-threatening. It requires premarket approval (PMA) to confirm safety and effectiveness of medical software. Examples include AI systems that diagnose a serious condition like a brain aneurysm from MRI scans.
The FDA has also put out a guidance called “Content of Premarket Submissions for Device Software Functions” in June 2023. This guide helps companies that develop SaMD better understand the documentation they will need to submit for medical devices. There are two levels of documentation that companies should prepare: basic (for software that does not pose a risk of death or serious injury if it fails) and enhanced (for software whose failure could likely cause serious injury or death before risk controls are applied).
Basic documentation includes a brief description of the workflows used to manage the software life cycle, including:
- Processes and procedures used in software development, verification, and validation.
- Software coding standards, methods, and tools used in software development.
- Key deliverables of activities and tasks associated with software development, verification, and validation.
- Processes, procedures, and tools used to link user needs, system requirements, software requirements, software design specifications, software testing, and implemented risk controls
- Processes and procedures used in software configuration and change management.
- Processes and procedures used in software maintenance, including software change risk assessment, initial testing that evaluates the correctness of implemented software changes, and regression analysis and testing.
Enhanced documentation includes all the elements of basic documentation plus additional, detailed information to demonstrate robust risk management and software assurance, including:
- Software description
- Risk management data
- Software requirements specification
- Software architecture design
- Software design specification
- Software development, configuration management and maintenance
- Verification and validation testing
- Software version history
- Unresolved software anomalies
Note: The same FDA guidance, “Content of Premarket Submissions for Device Software Functions,” states that instead of providing detailed documentation of every aspect of the software development, a manufacturer may choose to submit a “Statement of Conformity” (Declaration of Conformity) to ANSI/AAMI/IEC 62304 “Medical Device Software – Software Life Cycle Processes.” IEC 62304 covers the entire software lifecycle of medical devices that meet FDA expectations for software quality and safety.
SaMD regulations and classifications in the EU
In the EU countries, there are two regulations for medical devices: MDR (Medical Device Regulation) for general medical devices that classified software as Class I, IIa, IIb, III and IVDR (In Vitro Diagnostic Regulation) for in vitro diagnostic devices (e.g. laboratory software for blood analysis) with Class A (low risk), B( moderate risk), C (high risks), D (higher risk), where:
| EU Class | Risk | Purpose | Regulatory path |
|---|---|---|---|
| Class I | Low-risk software | For health tracking applications | Self-certification for CE mark |
| Class IIa | Medium-risk software | For monitoring physiological processes | Notified Body |
| Class IIb | Higher-risk software | For diagnostic diseases that influence significant treatment decisions | Notified Body |
| Class III | High-risk software | For critical or life-threatening situations | Notified Body |
The class depends on:
- the intended use of the device
- the risks to the patient associated with its use
Sometimes the EU classification does not fully reflect the risks of medical software, so the International Medical Device Regulators Forum (IMDRF) classification is used as additional guidance. It also classifies medical software based on its purpose and the level of risk to the patient but identifies four categories of medical devices (I, II, III, IV). To help you identify what category your device falls under, just look at the table below.
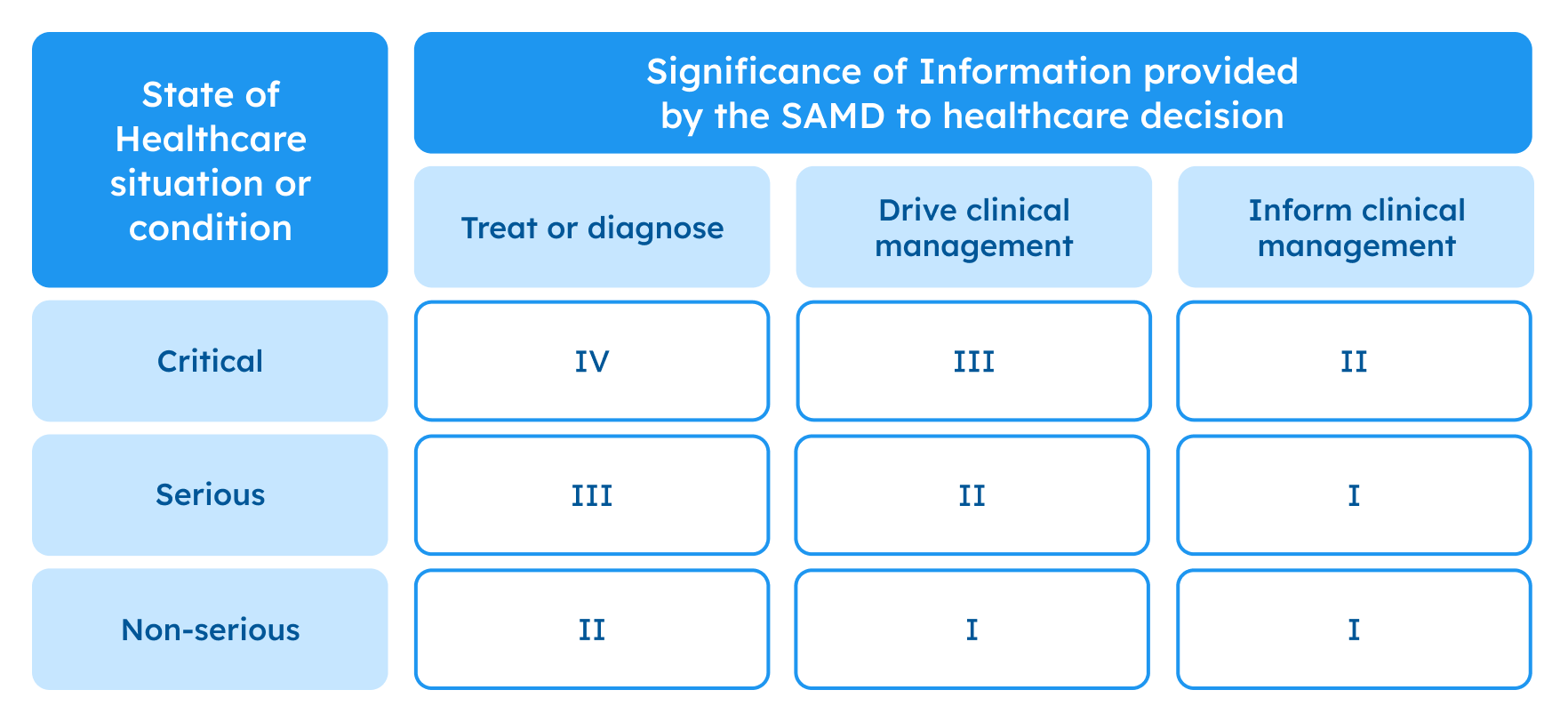
Possible framework for risk categorization and corresponding considerations, IMDRF. Source
To make it, you can use MDCG 2019-11 – an official EU document where on page 27 you can find instructions for software classification and Annex IV that contains examples of SaMD classification using the IMDRF, but adapted to European risk classes (Class I, IIa, IIb, III).
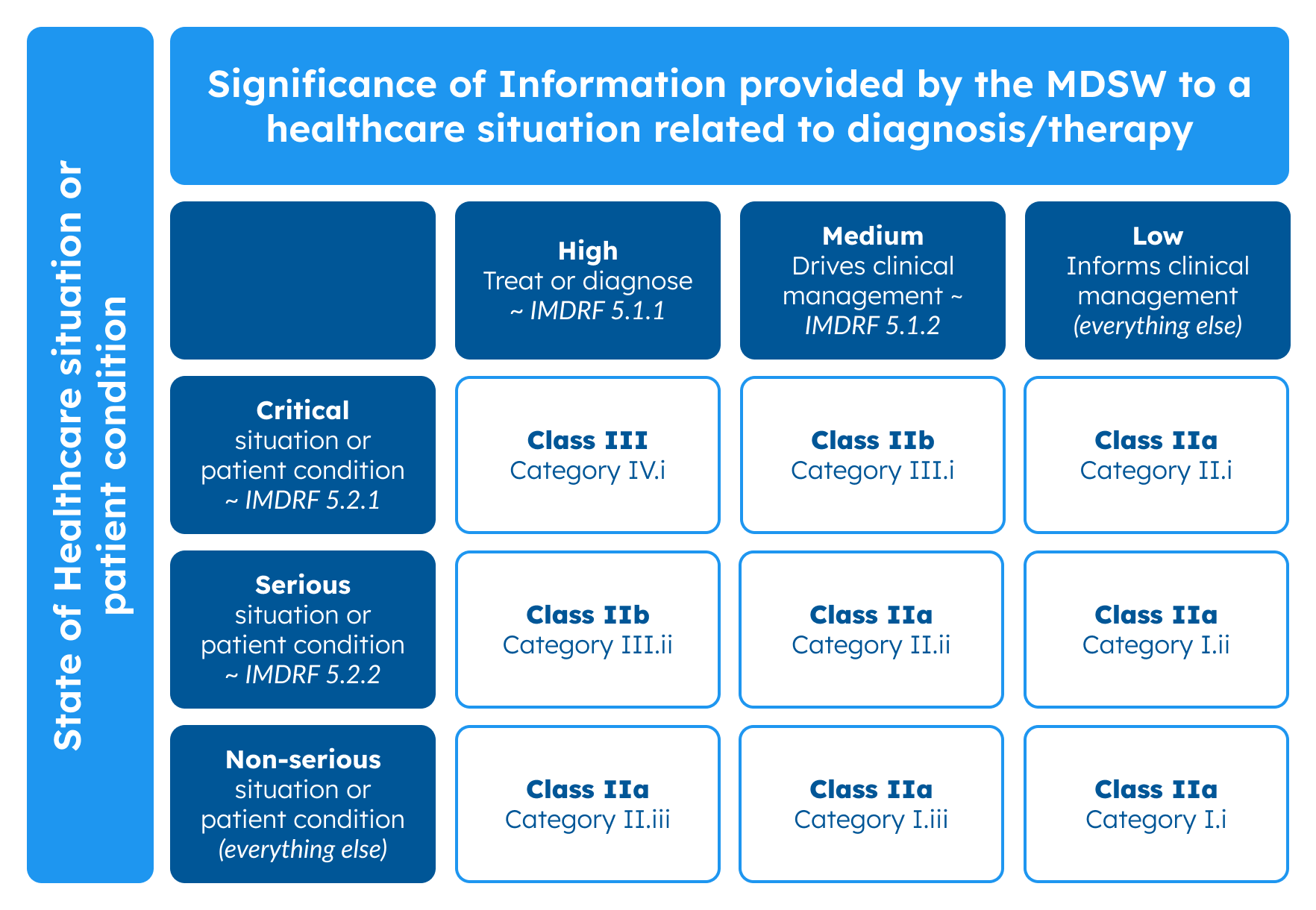
Classification guidance on rule 11, MDCG 2019-11. Source
The higher the medical software risk and class, the more stringent the certification and documentation. Class I can be certified through self-certification, while Class IIa–III requires the involvement of a Notified Body (EU) and FDA 510(k)/PMA (US).
Similar to the FDA approach, EU regulators expect comprehensive technical documentation that covers:
- Software lifecycle management (aligned with IEC 62304)
- Risk management (aligned with ISO 14971)
- Quality management (aligned with ISO 13485)
- General requirements for product safety (aligned with IEC 82304-1)
- Application of risk management for IT-networks incorporating medical devices (IEC 80001)
- Application of usability engineering to medical devices (IEC 62366-1)
- Verification and validation evidence
- Clinical evaluation and software testing
- Post-market surveillance and maintenance plan
Software safety classification according to IEC 62304
When you develop Software as a Medical Device, you must comply with specific regulations listed above. One of the key international standards is IEC 62304. It defines how software should be developed, maintained, and managed throughout its lifecycle. The key idea of the standard is a software safety classification that indicates how strictly software should be developed based on the risk to the patient. IEC 62304 defines three software safety classes:
- Class A: no injury or damage to health
- Class B: non-serious injury
- Class C: serious injury or death
This classification is not a determination of how safe your software is. Instead, it is the basis for determining how rigorous your software development process must be. You should keep in mind that your safety classification under IEC 62304 does not directly correspond with risk classification under US or EU regulations. For example, if your software has a safety classification class C, then there’s a good chance your device will be class III (for either the US or the EU).
Medical device software development principles
To successfully bring SaMD to market and ensure its long-term success, companies need to follow a structured software development approach.
Risk classification
At this stage, you need to define the SaMD objective and assess the potential patient harm it poses. Next, you need to select the system classification by region (FDA in the US or MDR in the EU) and determine the software risk level. This will help you define regulatory oversight requirements. Also, you need to classify software by safety risk (Class A/B/C) according to IEC 62304 to identify the level of rigor required for software development, testing, and verification. Proper software risk classification ensures that security controls, verification processes, and regulatory measures are appropriate to the potential damage in the event of a software failure.
Software Development Lifecycle (SDLC)
Once the risk classification is determined, the next step is to define and implement a robust software development life cycle (SDLC). According to IEC 62304, the software development process begins with planning and ends with its maintenance, always accompanied by detailed documentation. Between these stages, several necessary steps must be completed, including:
- Software requirements analysis: define functional, non-functional, and regulatory requirements to ensure software meets user needs, healthcare goals, and applicable standards.
- Software architectural design: create software architecture and design specifications, including data flows, user interfaces, and integration points, to ensure a safe and efficient implementation.
- Software unit implementation and verification: develop individual software units (modules) and verify that each performs correctly according to specifications.
- Software integration and integration testing: combine software units into a complete system and test their interactions to verify correct operation.
- Software system testing: validate the entire system against requirements to ensure it functions safely and reliably in its intended environment.
- Maintenance and updates: monitor performance, fix bugs, and implement updates to maintain compliance, reliability, and patient safety during software use.
Clinical evaluation
Clinical evaluation is a planned, continuous process for collecting and analyzing medical data about a SaMD. Its goal is to confirm that the software’s output is clinically relevant and that software performs as intended in real medical use. In general, this process ensures that SaMD delivers reliable, predictable results for patients and physicians. The clinical evaluation of SaMD, as described in the IMDRF guidance adopted by the FDA, includes three main pillars:
Valid clinical association: answer the question “ Is there a valid clinical association between your SaMD output and your SaMD’s targeted clinical condition?”. You must demonstrate that the intended use of the SaMD is clinically relevant and supported by medical evidence. Examples of evidence include:
- Literature searches
- Original clinical research
- Professional society guidelines
- Secondary data analysis
- Performed clinical trials
Analytical validation: answer the question “Does your SaMD meet technical requirements?” You must evaluate the technical performance and data processing accuracy of your software. Generate evidence during verification and validation activities as part of your software development practices.
Clinical validation: answer the question “Does your SaMD generate clinically relevant outputs?” You must demonstrate that the use of the software results in clinical effectiveness and outcomes consistent with the intended medical purpose. Show clinical significance using evidence collected during verification and validation activities. Examples may include:
- Existing studies that support the same intended use
- Real-world clinical performance data
- Usability and human factors evaluation
This clinical evidence must be documented for regulatory submissions and CE marking to show that the SaMD is safe and effective.
Regulatory submission and approval
When developing a Software as a Medical Device, the regulatory submission and approval phase is critical to ensuring that the software can be legally marketed and used in clinical settings. Regulatory approval for SaMD comes down to three things: correct classification of the product, collection of a complete set of documentation, and compliance with the required filing procedure (own for each country). For the US (FDA), the filing type depends on the software intended and risk classification:
- 510(k) premarket notification: for moderate-risk devices (Class II) that are substantially equivalent to a legally marketed predicate device.
- De Novo request: for low- or moderate-risk devices without a predicate, providing a new classification for the device.
- Premarket Approval (PMA): for high-risk devices (Class III) requiring extensive clinical data of safety and efficacy.
For the EU (MDR/IVDR), manufacturers must prepare a technical documentation and work with a Notified Body to obtain CE marking, which demonstrates conformity with the Medical Device Regulation (MDR 2017/745) or In Vitro Diagnostic Regulation (IVDR 2017/746). The process includes:
- Classification of the device according to risk (Class I, IIa, IIb, III for MDR; Class A–D for IVDR).
- Compilation of technical documentation: basic or improved, which includes software design, verification, validation, risk management, clinical evaluation, quality management system compliance, etc.
- Assessment by the Notified Body (if required by class) before CE marking and market release.
At this stage, a robust quality management system (usually ISO 13485-certified) must be implemented to ensure control over all processes. Once approval or certification is obtained, SaMD software can be released to the market, but its lifecycle does not end there.
Post-market surveillance and maintenance
Once your SaMD is on the market, you should follow the post-market surveillance (PMS) process and maintenance. Post-market surveillance processes focus on monitoring the product in real-world conditions to identify potential risks and issues from intended performance after release. It involves systematically collecting user feedback, tracking complaints and incidents, and monitoring clinical data and usage logs.
Real-world performance monitoring is especially vital, as it helps companies detect issues such as algorithmic bias (for AI/ML-based software), unforeseen use cases, or rare failure modes, and then correct them. Any necessary software changes must comply with validation procedures and regulatory approval.
As for maintenance requirements, companies need to establish a software maintenance plan in accordance with IEC 62304. It usually covers functional software enhancements (bug fixes, updates), configuration and risk management, etc. Software maintenance plans should be part of the overall Quality Management System (QMS) and linked to post-market surveillance to address field issues. Effective maintenance requires coordination between IT, compliance, clinical, and quality teams to ensure software remains secure and reliable.
Rigorous Development Process
Developing software as a medical device is one of the most challenging areas of modern software engineering. Complex system integrations, compliance with a wide range of standards and regulations, attention to security – all of that should be implemented on the highest level, as it affects the health of real people. It means developers must adopt an end-to-end approach, incorporating security by design, conducting different types of testing, and continuous security monitoring. With proper planning and implementation of the SaMD system, the client, together with the development team, not only advances healthcare quality through technology but also enhances patients’ quality of life worldwide.
Challenges and solutions when building SaMD
While SaMD offers notable benefits, it also presents challenges that manufacturer companies must be aware of to address or avoid them successfully.
Interoperability
Often, SaMD solutions integrate with other systems, like EHRs, medical devices, lab platforms, and cloud services. This integration can be complex because various solutions may differ in data formats, communication protocols, and regulatory requirements, hindering seamless integration.
Solution: To overcome interoperability issues, companies need to use standard communication protocols (FHIR, HL7, DICOM), implement standardized data formats (like HL7 FHIR for healthcare data), and ensure compliance with relevant regulatory frameworks. Also, they can apply middleware to handle protocol translation and conduct thorough testing in various integration scenarios to ensure compatibility with diverse third-party systems.
Data security and privacy
Because SaMD software handles sensitive patient data and can be connected to networks, devices, and cloud services, it is extremely vulnerable to cyberthreats. Hacking or malware penetration can not only compromise sensitive data but also potentially disrupt software that leads systems’ downtime and potentially harms the patient.
Solution: to mitigate cybersecurity and privacy risks when developing a SaMD solution, developers should implement a multi-layered security strategy, including end-to-end data encryption, role-based access control with proper authorization, multi-factor authentication, and real-time system monitoring. They should conduct regular vulnerability assessments, penetration testing, timely software updates and develop an incident response plan. Moreover, companies should follow industry regulatory standards (e.g., FDA, ISO 13485, IEC 62304) from the ground up to ensure software compliance and resilience during and after development.
Data integrity and reliability
SaMD software processes data from multiple sources, such as images, wearable devices, laboratory systems, and electronic medical records, whose quality and format can vary significantly. This data diversity creates the risk of inaccurate, incomplete, or inconsistent information, arising from software failures, network disruptions, or human errors. A result of this is inaccurate clinical decisions, misdiagnosis, or inappropriate treatment recommendations, all of which directly impact patient safety.
Solution: To ensure data accuracy and consistency during SaMD development, companies should use data standardization to convert incoming data from various sources into consistent formats, and validation techniques to verify its correctness and reliability. They need to conduct regular data backups to protect information from system failures and data audits to identify and rectify serious issues before they escalate. Also, companies need to implement security monitoring mechanisms and establish a comprehensive security policy to safeguard sensitive information throughout the software lifecycle.
Performance and reliability
After deployment in a healthcare environment, SaMD operates in real-world conditions that may differ from the controlled environment in which it was developed. SaMD performance and reliability issues may arise due to limited connectivity or device computing power, or from system overload during peak usage. Any delays or disruptions in software operation could lead to delayed diagnosis or reduced clinical effectiveness, posing risks to patient safety.
Solution: To ensure consistent software performance, companies should use load balancing strategies and conduct stress/performance testing before the release. They also need to implement monitoring systems to track uptime, latency, and processing accuracy in real time, as well as redundancy mechanisms to maintain operation even if connectivity is lost. Performance requirements should be defined upfront and continuously verified throughout the device lifecycle.
Real-life examples of medical device software
Modern healthcare companies already employ SaMD products that bring numerous benefits to users around the globe. Below are a few notable examples of them.
ECG analysis software
The company AliveCor developed Kardia devices as personal ECG (electrocardiogram) monitors for tracking heart health on the go or at home with great accuracy. Users place their fingers on the device’s electrodes, which measure the heart’s electrical activity and transmit the data to a smartphone via Bluetooth. The AliveCor app analyzes the data using AI, displays the ECG, and identifies any heart rhythm abnormalities. Users can view the results on their phone and share them with a doctor for interpretation.
Also, the application stores heart measurements over time and provides detailed reports and monthly summaries. This enables patients to continuously monitor their heart health, reduces the need for frequent in-person check-ups, and enables timely treatment.
Sleep diagnostics software
The company Covidien (later acquired by Embla) introduced Sandman Elite, a sleep diagnostic software platform used in sleep labs to assess patient sleep quality and enhance sleep disorder diagnostics. The system collects and converts physiological signals, such as brain waves (EEG), heart rate, and respiration, into digital form in real time. After that, it analyzes these signals to help researchers interpret sleep study results and generate detailed reports for clinical and research use.
Glucose monitoring system
The company Medtronic introduced the MiniMed 780G glucose monitoring system, which combines glucose monitoring (CGM) sensors with AI to provide real-time glucose data, predictive alerts, and automatic insulin dose adjustments. The system uses the sensor, which measures glucose levels and transmits data wirelessly to an insulin pump. Equipped with SmartGuard technology, the pump automatically adjusts insulin delivery based on glucose readings. Users can track glucose levels, receive alerts about high or low glucose levels, and view insulin dosing information via a mobile app.
Cognitive behavioral therapy software
The company Feel Therapeutics developed a cognitive behavioral therapy (CBT) software designed to improve mental health for people with depression, anxiety, and sleep issues. It collects data on mood, stress, sleep, activity, and cognitive function in real-time. Then, it analyzes this data and, based on the results, selects programs and recommendations on how to support emotional well-being and improve stress management skills. The solution applies scientifically proven methods such as cognitive behavioral therapy (CBT), mindfulness, and positive psychology.
Users can take interactive classes, watch short educational videos, and connect with qualified coaches. The app also allows them to keep an emotion diary and integrates with the sensor for more precise personalization.
The future of SaMD solutions
As new technologies develop, and medical regulations and standards improve, the need for Software as a Medical Device (SaMD) will only grow. In the future, we’ll most likely see the rise of digital therapeutics software for personalized treatment and the integration of remote monitoring and telemedicine solutions for ongoing, remote patient care. We will also see the use of IoT for more comprehensive health monitoring, AI for improved diagnostics, and sensors with digital biomarkers for more personalized treatment planning.
Now, Software as a Medical Device is transforming modern healthcare by enabling more accurate diagnostics, proactive health monitoring, and personalized treatment without traditional medical equipment. However, to successfully bring such a solution to market, companies should ensure regulatory compliance, correctly classify risks, implement a structured software development lifecycle, and conduct rigorous post-market monitoring.
If you need a trusted provider for reliable medical software development, contact SoftTeco. Our experts will provide you with compliant Software as a Medical Device built to the highest industry and security standards – all in line with your requirements.
FAQ
How can I determine that my project meets the requirements for SaMD?
– Does your project use software?
– Is your software used for medical purposes (to diagnose diseases, treat or prevent diseases, monitor health, or analyze medical data for health decision-making)? If at least one of the following items is checked, then yes.
– Can your software be used without being part of a specific hardware device?
If you answer “yes” to some questions, then your project uses the SaMD model. If the answer to any of these questions is “no”, then the project does not meet the SaMD requirements.



Comments